Abstract
AUTS2-Related Neuropsychiatric Disorders with Intrafamilial Variability
Intellectual disability, autism and psychiatric conditions are a group of clinically and genetically heterogeneous neurodevelopmental disorders. Their pathogenesis involves many genes responsible for neuronal migration, axon extension, synaptic function and transcriptional regulation. Among them, autism susceptibility candidate 2 (AUTS2, OMIM *607270) gene has been recurrently associated with syndromic intellectual disability and autism: both single nucleotide and intragenic exonic deletions have been reported. Genotype-phenotype correlation studies suggest that C-terminal disruptions have a more severe phenotype with respect to N-terminal ones.
We report on a family segregating and intronic variant with functional effects in AUTS2, showing marked intrafamilial variability at the clinical level in three carrier members: the proposita, a 24-years-old girl with developmental delay, severe hyperactivity, sleep apnoea, support teacher from early on and self-harmful behaviours; her 20-years-old brother with hyperactivity, cognitive impairment and developmental motor delay; the mother, who displays peculiar facial features, strikingly resembling those reported in AUTS2 patients, and referred a support teacher at school.
Intronic deletions in AUTS2 segregating with the known phenotype are not acknowledged as all the reported deletions encompass exons: therefore, the deletion observed in our family might be crucial to identify new regulatory sequences. The patient’s deletion, even though it is located far from the intron-exon boundary, includes two CpG islands and one regulatory element, whose removal might impair the appropriate expression of AUTS2. Besides that, RNA studies confirmed an exon skipping resulting in the loss of eighteen aminoacid residues in the N- terminal region.
Author(s):
Abstract | Full-Text | PDF
Share this

Google scholar citation report
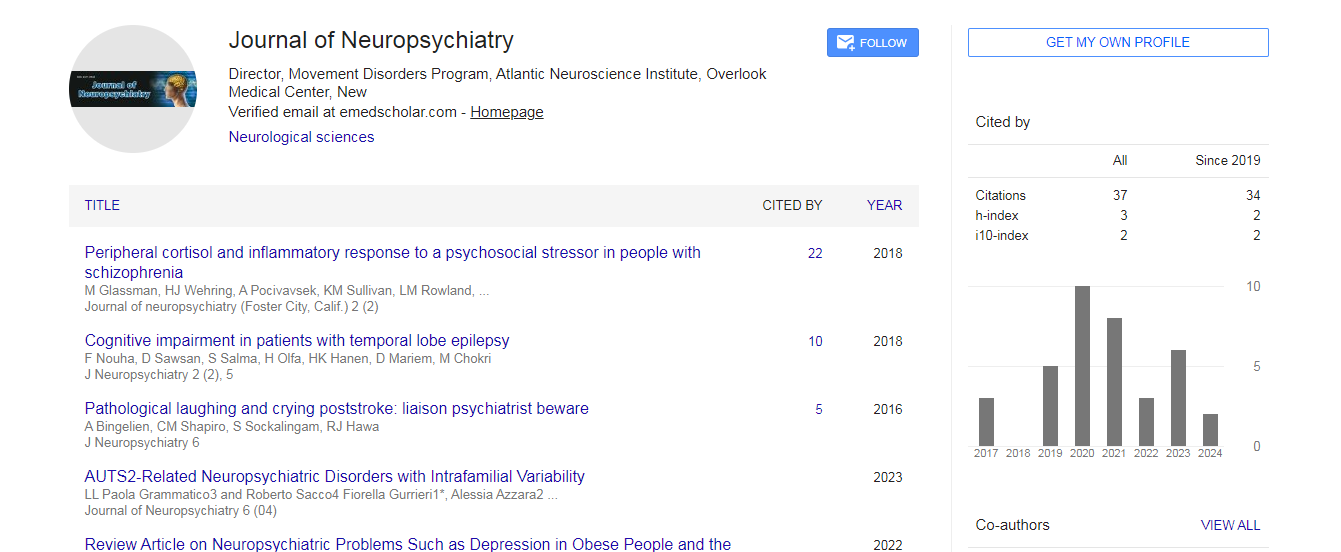
Citations : 37
Journal of Neuropsychiatry received 37 citations as per google scholar report
Abstracted/Indexed in
- Google Scholar
- China National Knowledge Infrastructure (CNKI)
- Secret Search Engine Labs
- Euro Pub


Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences